- Biography
- Works
- Contact
The information below refers to the time of the award.
A German and Danish resident, Matthias MANN was born in 1959 in Lingen (Germany). He studied physics and mathematics at the University of Göttingen (Germany), and received his doctorate in chemical engineering from Yale University (United States), under the supervision of John B. Fenn, winner of the Nobel Prize in Chemistry. On his return to Europe in 1989, he first worked as a post-doctoral Fellow then as a Senior Scientist in the Department of Molecular Biology at the University of Southern Denmark in Odense, before joining the European Molecular Biology Laboratory (EMBL) in Heidelberg, where he directed the Protein & Peptide Group. In 1998, he returned to Odense as a full Professor of Bioinformatics in the Department of Biochemistry and Molecular Biology at the University of Southern Denmark. Since 2005, he has held the position of Director of the Department of Proteomics and Signal Transduction at the Max-Planck-Institute of Biochemistry in Martinsried (Germany). In 2007 he was additionally appointed as Director of the Department of Proteomics, at the Novo Nordisk Foundation Centre for Protein Research, Faculty of Health Sciences, University of Copenhagen, Denmark.
Matthias MANN has authored or co-authored more than 440 publications, making him one of the most highly cited researchers worldwide. A member of EMBO (European Molecular Biology Organization) and of the Royal Danish Academy of Sciences, Matthias MANN has received numerous distinctions, particularly the Lundbeck and the Novo Nordisk Research Prizes, the Meyenburg Cancer Research Award, the Schelling and the Leibniz Prizes.
Mass spectrometry meets the proteome
After the genome comes the proteome. Having decrypted the genetic heritage of numerous species including humans, biologists are now aiming at characterizing the totality of the proteins in a cell or an organism. Technically, this is an extremely difficult task.
Matthias MANN progressively succeeded in overcoming the obstacles and turned mass spectrometry (an analysis technique already widely used for detecting and measuring the structure of molecules by measuring their mass) into a highly effective tool for characterizing the proteome.
With his colleagues, he managed to extract the proteins from the gel used by biologists to separate these molecules, but which previously rendered analysis via mass spectrometry impossible. He then miniaturized electrosprays, a technique used for ionizing molecules, and thus considerably enhanced the sensitivity of the analysis. Lastly he used mathematical algorithms to identify protein fragments by comparing them with those already listed in databases. Thanks to this work, biologists can now routinely use mass spectrometry for the study of proteins.
Matthias MANN then built on this progress to achieve another step forward by developing the new and highly accurate SILAC method (Stable Isotope Labelling by Amino Acid in Cell Culture). This technique, which characterises the functions of proteins, has opened the way to many applications of proteomics.
The proteome
Proteins are essential functional entities in cells. For example, structural proteins determine the architecture of tissues while enzymes regulate chemical reactions. It is therefore logical that having focused their attention on genes and on decrypting the genome of numerous species including humans, biologists are now aiming at characterizing the totality of the proteins in a cell or an organism, known as its proteome. Proteomics, a new branch in biology, is emerging with the objective of achieving reliable quantitative analysis of individual and combined proteins.
The biologists needed an effective tool to achieve this. Mass spectrometry immediately came to mind. This technique is widely used to detect and define molecule structure by measuring their mass, but initially it was thought to be inappropriate for protein analysis since, unlike DNA, proteins cannot be amplified. This fact, combined with that of the great complexity of high and low abundance proteins in biological systems, made the approach difficult to implement.
One by one, Matthias Mann’s group overcame the obstacles to protein analysis by mass spectrometry. First of all they managed to extract the proteins from the gel which was the form in which biologists always had their proteins and which rendered analysis via mass spectrometry impossible. They then miniaturized electrosprays, a technique used for ionizing molecules under study, and developed the “nano-electrospray” which considerably enhanced the sensitivity of the analysis. And lastly they used mathematical algorithms to identify protein fragments by comparing them with those already listed in databases.
Matthias Mann then achieved another step forward by developing the new and extremely accurate SILAC (Stable Isotope Labelling by Amino Acid in Cell Culture) method for proteome quantification. SILAC has opened the way to many ingenious applications of proteomics to characterize the function of proteins via their interactions, modifications and cellular localizations.
Matthias Mann’s work has revolutionized proteomics. Biologists can now use mass spectrometry for the study of all kinds of proteins – whether of bacterial or human origin.
The contribution of mass spectrometry
The proteomic workflow (Fig. 1) developed by Matthias Mann and his colleagues involves first solubilizing the proteins in strong detergents, then digesting them to peptides (smaller combinations of amino-acids). They are then separated in very narrow columns (75 micrometer inner diameter) and ionized by electrospray. In the mass spectrometer, the peptide masses and intensities are then measured very accurately.
The researchers then sequence the peptides by colliding them with gas and recording the mass spectrum of the resulting fragments – a procedure called MS/MS. The resulting data are analysed using MaxQuant, a computational proteomics platform developed by Matthias Mann’s team and freely available to the scientific community. The end result is a list of proteins quantified according to different cellular states. For example, Matthias Mann’s team succeeded in analysing a substantially complete proteome in the haploid and diploid states of yeast.
Applications to proteins post-translational modifications
Proteins are the functional entities in cells. However to fulfil these functions, proteins often require post-translational modifications such as attachment of phospho-groups to proteins (phosphorylation), of small proteins like ubiquitin or any of the hundreds of other modifications that are known so far. The technology developed by Matthias Mann can also be applied to detect and quantify these protein modifications. One of the big surprises of the last years has been just how numerous these modifications are. In a mammalian cell Matthias Mann’s team can now use mass spectrometry to detect more than 10,000 phosphorylations or ubiquitinations with single amino acid accuracy. The challenge for the proteomics and biological communities is now to determine the functional roles of these many and diverse modifications.
Applications to protein interactions
Quantitative proteomics methods can also be applied to determine with great precision the protein interactions that play an important part in many cellular processes. By incubating a cellular proteome with the protein under study (bait) and measuring the proportion of proteins that interact with the bait and with the beads to which they are attached, even weak and transient interacting proteins can be detected. Matthias Mann’s group has undertaken a large-scale experiment with high confidence levels to determine the network of protein interactions within a cell.
Interactions of any bait molecule with proteins can also be determined by the same technology. Matthias Mann and his colleagues have studied the interactions of epigenetic marks with protein complexes that read them. These technologies can also be used to determine the proteins that bind to specific DNA sequences, which can help to unravel the functional mediators of genetic differences between individuals of a given species.
Implications for the diagnosis and treatment of cancer
The work completed so far has considerably simplified the task of today’s proteomics specialists, but it could also have diagnostic and therapeutic applications, particularly with regard to cancer.
Recent research conducted by Matthias Mann’s team now allows the quantitative analysis of human tumour material as well. The researchers showed that mass spectrometric analysis can be used on biological samples, even formalin-fixed, paraffin embedded material, revealing not only the precise quantity of thousands of proteins, but also how they are modified.
This opens up the possibility to find biomarkers for the different phases of a cancer that could guide diagnosis and treatment. Preliminary work undertaken by Matthias Mann’s group has already shown that proteomics data can easily distinguish between different types of B-cell lymphoma that are indistinguishable by traditional histological means. Likewise, the methods developed by the German research team now make it possible to analyse the tumours of dozens and hopefully soon, hundreds of patients with breast cancer or prostate cancer (Fig. 2). There is reason for hope that Matthias Mann’s methods will contribute to improving the classification of patients and will be helpful in deciding optimal treatment.

Fig. 1 – Workflow of high-resolution, quantitative proteomics.
The different stages involved in the use of mass spectrometry for proteome analysis. This example shows the response of the cellular proteome to short term growth factor stimulation (Adapted from Cox J. and Mann M., Is proteomics the new genomics?, Cell, 2007, 130 (3): 395-8, with permission).
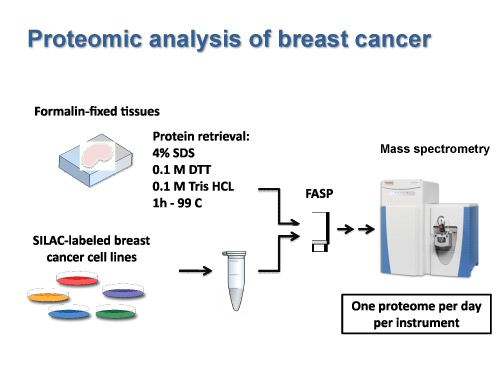
Fig. 2 – Proteomic analysis of breast cancer
The tumour biopsies fixed in formalin and embedded into paraffin (FFPE) are prepared using the FASP method (Filter Aided Sample Preparation). This allows for the extraction and solubilisation of the proteome and digestion into peptides. (Tamar Geiger, Jacek Wisniewski and Matthias Mann).
Professor Matthias Mann
Director
Department of Proteomics and Signal Transduction
Max-Planck Institute of Biochemistry
Am Klopferspitz 18
82152 Martinsried, Germany
Tel.: +49 (0)89 8578 2558 (PA) 2557 (direct)